 CONTACT US
CONTACT US
First Principles Study of the Interfaces
First-Principles study of the Interfaces
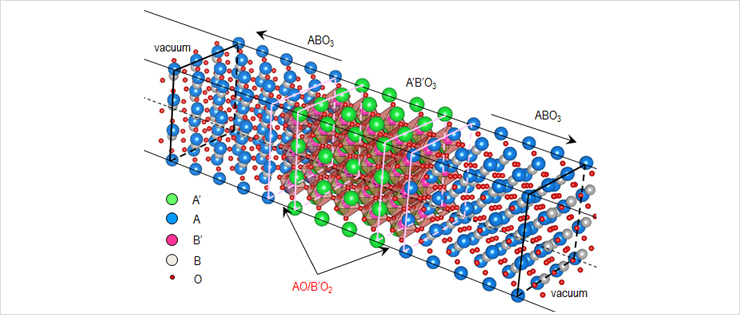
A central issue in controlling the novel behavior in oxide heterostructures is to understand how various physical variables (spin, charge, lattice and/or orbital hybridization) interact with each other.
In particular, density function theory (DFT) has provided significant insight into underlying physics of materials at the atomic level, giving quantitative results consistent with experiment.
Using density functional theory methods, we explore the electronic, magnetic and structural properties developed near the interface in various oxides (SrTiO3/LaAlO3, EuO/LaAlO3, Fe/PbTiO3/Pt, Fe//BaTiO3/Pt and Cs/SrTiO3 so on) heterostructures. We study the interplay between physical interactions, and quantify parameters that determine physical properties of hetetrostructures. These theoretical studies help understanding how physical variables couple with each other and how they determine new properties at oxide interfaces.
Busandaehak-ro 63 beon-gil, Geumjeong-gu, Busan 609-735, Korea, TEL:82-51-510-2227
Copyright by Pusan National Univ. CPMD Lab. All Rights Reserved.